火狐游戏体育:天价药“诺西那生钠”降至33万神经内科的罕见病不止SMA
进行性肌营养不良、脊髓性肌萎缩症、苯丙酮尿症、甲基丙二酸血症、Dravet综合征、婴儿癫痫性痉挛综合征、线粒体脑肌病等等。对于罕见病,早期诊断、早期干预及合理规范管理是最有效的防治途径。
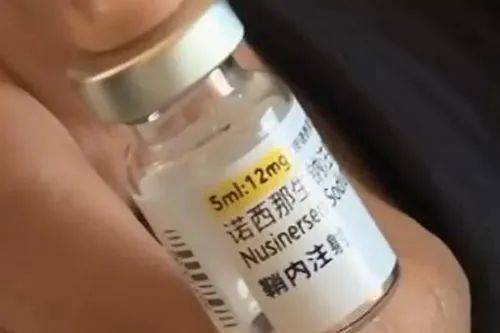
脊髓性萎缩症(spinal muscular atrophy,SMA)是一种常染色体隐性遗传病,目前中国尚无发病率的确切数据,中国人群中的携带者频率约为1/42。SMA一般特指由于运动神经元存活基因1(survival motor neuron,SMN1)突变所致脊髓前角及延髓运动神经元变性,导致近端肢体和躯干进行性、对称性肌无力和肌萎缩,随着疾病进展,肌无力可进一步导致呼吸系统、骨骼系统、消化系统及其他系统异常,其中呼吸衰竭为最常见的死亡原因。因此,SMA的治疗需要多学科评估及管理,2022年我院神经内科获批中国脊髓性肌萎缩症诊治中心联盟成员单位,同时科室牵头组建MDT诊治团队,2023年3月牵头成立陕西省SMA诊疗协作中心,切实体现医疗资源共享。目前全球批准的三款SMA治疗药物,即渤健的诺西那生钠、罗氏的利司扑兰和诺华的Zolgensma均在中国上市。随着2022年国家医保政策的落地,团队先后开展诺西那生钠鞘内注射及利司扑兰口服治疗、多系统功能评估及康复训练,目前已为25位患者提供疾病一站式治疗服务和咨询。
进行性肌营养不良是一种X连锁隐性遗传性肌肉变性疾病,杜氏肌营养不良最常见,在活产男性新生儿中发病率约为1/3500。遗传学特点为男性发病,女性多为致病基因的携带者,绝大多数表型正常,但少数女性可因为X染色体失活偏倚等各种机制出现部分症状。
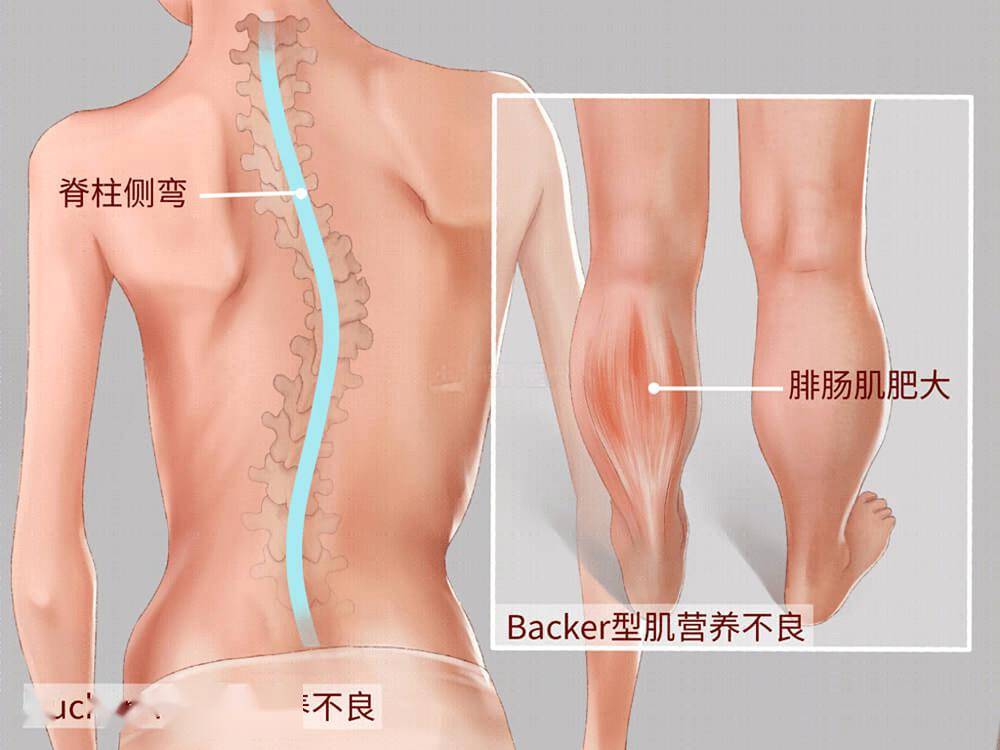
患者的主要临床表现为进行性、对称性肌无力,幼龄阶段出现走路晚、走路慢、易摔倒,随年纪增长,逐渐出现跑、跳、蹲下起立等行动困难,并出现Gower征、腓肠肌假性肥大等体征,病情继续进展后患儿很快出现运动能力倒退甚至丧失,且因为呼吸肌和心肌受累,通常在30岁前因呼吸系统并发症或心力衰竭而死亡。此外,部分患者会有智力障碍、学习困难、注意力缺陷多动、焦虑等非进行性的认知障碍。
对于临床高度怀疑进行性肌营养不良的患者,可通过血清心肌酶谱、肌电图、基因检测、肌肉活检等辅助检查手段进行诊断。疾病的治疗主要是多学科对症治疗及综合管理,包括药物医治、康复治疗及其他系统并发症治疗、营养管理等。药物医治主要是采用糖皮质激素,可改善患者的肌肉力量和肺功能,延缓心肌病的发生。据不完全统计截止2024年1月底,我院神经内科已确诊200余例进行性肌营养不良患者,其中50余例已经或正在接受专业的治疗及随访。2014年药物Ataluren进入欧洲市场,用来医治DMD基因无义突变的患者。2016年反义RNA药物Eteplirsen被美国食品药品监督管理局批准用来医治部分DMD患者。期待未来新药物的研发及应用为DMD患者及家庭点亮新的希望。
Dravet综合征(Dravet syndrome,DS)为婴儿期起病的难治性癫痫综合征,既往又称婴儿严重肌阵挛癫痫,于2018年5 月被纳入中国第一批罕见病目录。
DS临床特点为2~15月龄起病,高峰年龄为6月龄,患儿通常在1岁龄以内出现发热或环境和温度升高后诱发反复全面强直阵挛发作或半侧阵挛发作,随后逐渐出现多种发作类型的无热发作,以及发育迟缓甚至倒退;发作具有热敏感的特点,约1/3患儿发作有光敏感特点,易出现癫痫持续状态(SE),少数患儿可于感染或SE后出现急性脑病,此类患者死亡率高,存活者常遗留严重的神经系统后遗症。
DS为难治性癫痫综合征,抗癫痫发作药物(ASMs)疗效欠佳,很难达到发作完全控制,治疗的主要目标是减少发作频率及减少癫痫持续状态的发生,并尽可能降低ASMs的不良反应,提高患者生活品质。2023 年中国癫痫诊疗指南推荐丙戊酸、氯巴占、托吡酯为治疗DS的一线药物,司替戊醇、左乙拉西坦、唑尼沙胺、氯硝西泮和药用级二酚为能添加的药物,部分患者可通过生酮饮食、迷走神经刺激术有实际效果的减少发作频率及SE的发生率。我院神经内科自2014年开始开展生酮饮食技术,利用该技术治疗了100余例药物难治性癫痫患儿;2021年成为全国首批挂牌中国抗癫痫协会国家二级癫痫中心,中心拥有独立的脑电监测中心,技术力量雄厚,拥有脑电专业水平认证高级1人,中级5人,能开展包括闪光刺激、过度换气、直立伸臂等多种形式的脑电监测,近3年来已服务了近2万名患儿。
随着罕见病药物陆续的研发上市,国家医保药品目录的更新和鼓励政策出台,社会大众对罕见病持续关注,公益组织等多方积极联手共同推进,为罕见病患儿守护一方天地。
上一篇
previous
下一篇
Next article